Lets face it there aren’t too many HTS pipelines that don’t touch a SAM/BAM files somewhere along the way. The name SAM comes from the acronym Sequence Alignment/Map format and they are really nothing more than tab delineated text files describing mapping information for short read data. Of course with the millions of reads generated by next gen sequencers these days the files become really really big. That is where BAM files come in, these contain the same information as SAM files but are encoded in condensed computer readable binary format to save disk space.
If you open up a SAM file in a text editor (or use “head” in bash) the first part of the file typically contains lines beginning with AT (@) symbol, which represents the “header”. The header contains general information about the alignment, such as the reference name, reference length, MD5 checksum, the program used for the alignment, etc etc. Every line after the header represents a single read, with 11 mandatory tab separated fields of information. Lets see how this works with a really simple example.
What I’ve done is mapped four ‘pairs’ of reads to the alcohol dehydrogenase gene (my favourite gene) using bowtie2. The figures below shows the stranded nature of the read pairs as well as where they map on the gene.
I have set this up so that the reads fall into these three groupings.
- Two pairs of high quality reads that concordantly map
- A read pair where only one read is high quality
- An orphan read (the R1 read is only 1 nt long aka trimming)
The next figure describes each of the mapping scenario’s using the colour encoding. The red set of reads behave as we would expect for paired end data.

Where the reads map and the different read scenarios
For the alignment I’m going to use bowtie2, all of the sequences are here if you want to do this yourself.
#index the reference and specify the prefix as reference
bowtie2-build reference.fa reference
#map the reads
bowtie2 reference -1 test_R1.fq -2 test_R2.fq > adh.sam
####bowtie output###
Warning: skipping mate #1 of read 'HWI-7001326F:39:C7N3UANXX:1:1101:10000:62296/1' because length (1) <= # seed mismatches (0)
Warning: skipping mate #1 of read 'HWI-7001326F:39:C7N3UANXX:1:1101:10000:62296/1' because it was < 2 characters long 4 reads; of these: 4 (100.00%) were paired; of these: 1 (25.00%) aligned concordantly 0 times 3 (75.00%) aligned concordantly exactly 1 time 0 (0.00%) aligned concordantly >1 times
----
1 pairs aligned concordantly 0 times; of these:
0 (0.00%) aligned discordantly 1 time
----
1 pairs aligned 0 times concordantly or discordantly; of these:
2 mates make up the pairs; of these:
1 (50.00%) aligned 0 times
1 (50.00%) aligned exactly 1 time
0 (0.00%) aligned >1 times
87.50% overall alignment rate
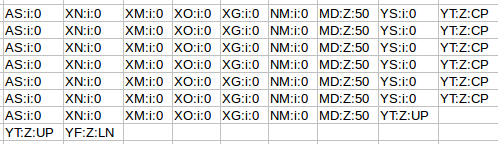
The “adh.sam” file can be opened with any text editor, but for most of this post I’m going to use the spreadsheet version just to easily work with all the columns. Note the header lines indicated with the “@”. In this case the @SQ relates to the reference and @PG relates to the program parameters. Each header line has fields in the format tag:value. For example, the @SQ line has SN (SN=reference sequence name) and LN (LN=reference sequence length). A full set of these header can be found in the SAM specs.
@HD VN:1.0 SO:unsorted @SQ SN:gi|568815594:c99321442-99306387 LN:1890 @PG ID:bowtie2 PN:bowtie2 VN:2.1.0
Below the header each read is represented by a line, it is worth nothing that even read “62296/1” which shouldn’t map since it is only 1 nt long also gets a line. Two other things to note are that firstly the reads can be ordered by reference coordinate, this is the default Tophat behaviour. Alternatively, they can be listed in pairs. Samtools can be used to switch between formats using the “sort” command and why does this matter? Some software needs to know how the reads are sorted, for example htseq-count expects reads ordered in pairs because read pairs can map different distances away from each other and finding them in the file increases memory requirements.
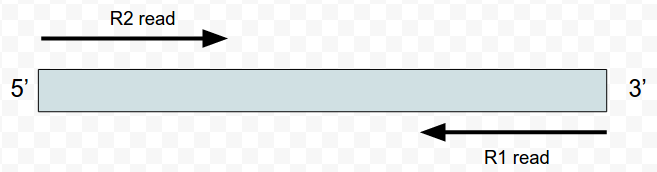
OK the fist four column. The QNAME (seq name) and RNAME (reference name) are pretty self explanatory. The FLAG uses information dense bit-wise encoding containing information on the alignment, such as the strand, if the pair mapped, if this if the first or second pair. If your dumb like me you’ll be thankful for the Broads has a nifty website that translates these numbers into plain English. For example, the 83 represents “read paired”, “read mapped in proper pair”, “read reverse strand”, “first in pair”. The 163 is “read paired”, “read mapped in proper pair”, “mate reverse strand” and “second in pair”. I think that makes sense. For reference the other two numbers are 137 (read paired, mate unmapped) and 69 indicating an unmapped read pair. Very cool. The final flag here POS corresponds to the alignment coordinates. We can confirm the reads are ordered R1 then R2 based on the mapping coordinates (see the figure at the top of the page).
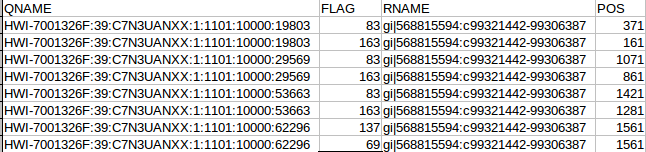
The next flags worth looking at are the MAPQ and CIGAR strings. The mapping quality is represented as a log transformed probability of mapping to the wrong position. Interestingly, the low quality (fastq quality “$”) has the same mapping score as the other high quality reads. I gather this is because the read really can only go one place when there is a 100% match and the reference is only short like this. The CIGAR string starts with a letter, in this case “M” for “alignment match” and 50 indicating the length of the alignment. The “*” indicates that no information is available. PNEXT is the position of the mate pair and TLEN is the observed template length, in other words the length of the sequenced fragment represented by the pairs.

Finally, we have sets of tags that provide other information on the alignment. Things like the number of ambiguous bases or gaps etc. Checkout the documentation of your aligner to find out what each tag defines if you are interested.