If you are looking for a lightweight but feature rich editor you can’t go past VIM! Sure those keyboard shortcuts are difficult remember and it’s frustratingly hard trying to stop reaching for that bloody mouse (the mouse will actually work in this set-up BTW). However, what I have found is the best way to learn VIM (I don’t think you ever really learn vim) is to make it useful enough that you want to start using it; this helps build up the muscle memory required to become efficient [enough]. The ultimate payback is that with practise you can become really really efficient relative to a user stuck with a mouse and complex GUI. The mouse is so 1980’s anyway 😛

So this post is about making VIM useful! The steps are based around Ubuntu/Debian Linux but should be adaptable for OSX and other NIX distros. Windows users, like yourselves I have no idea (-;?
Of course nothing I ever do is original and this especially goes for this post which borrows heavily from an excellent (but old) PyCon Asia talk by Martin Brochhaus. The .vimrc and much of the instructions are taken from the talk so feel free to watch it on youtube. I’ve also tried to update some of the info and stick to the minimum to get you coding away ASAP.
Section 1: Basic VIM install
These steps will clone the current version of VIM from source and enable additional features. If you already have a copy installed (ie vim starts when you type ‘vim’) I would first fun ‘apt-get remove vim’ (or your equivalent). First dependencies and some tools.
sudo apt-get update sudo apt-get install mercurial curl python-pip flake8 sudo apt-get build-dep vim #install python packages sudo pip install ipdb sudo pip install jedi sudo pip install flake8
As we are going to use a bin folder in our home directory we need to put this location into our path so that our new version of vim will run from the command line.
#add a home bin dir to your path nano ~/.bashrc #add the following to the end of the file if [ -d $HOME/bin ]; then PATH=$PATH:$HOME/bin fi #save and exit
Now we make the ~/bin dir as well as an ~/opt for the vim install. Eventually we will use a simlink from ~/bin (now in our path) to the ~/opt folders in our home directory, the -p flag says ‘don’t complain if this dir is already there’. The final command reloads your bashrc without you having logout. Cool.
mkdir -p ~/bin mkdir -p ~/opt source ~/.bashrc
Now to install VIM.
cd ~/Desktop hg clone https://vim.googlecode.com/hg/ vim cd vim/src ./configure --enable-pythoninterp --with-features=huge --prefix=$HOME/opt/vim make make install cd ~/bin ln -s ~/opt/vim/bin/vim #the following should return /home/YOUR_USERNAME/bin/vim which vim
Great!
Section 2: The vimrc settings file
Now we need a “.vimrc file”. This file will contain our customisations (the dot says I’m a hidden file). You can get mine from here (download to your desktop or copy contents into a blank txt file). It is worth look at this file in a text editor as it contains some information on the key short-cuts and the plugins we are going to use (BTW in .vimrc files a single quote ” indicates a comment (aka #) a double quote “” indicates a bit of code that can be uncommented to enable something). The next bit of code simply copies the file to your home dir and the gets a custom colour scheme (mostly just to show how to do it).
mv ~/Desktop/vimrc.txt ~/.vimrc mkdir -p ~/.vim/colors cd ~/.vim/colors wget -O wombat256mod.vim http://www.vim.org/scripts/download_script.php?src_id=13400
Section 3: Getting set-up for the plugins with pathogen
This next part is important. To manage plugins we will use a bit of kit called pathogen. The plugins can then be installed (mostly by a git clone) right into the “~/.vim/bundle/” (this will result in a folder structure like this: .vim/bundle/plugin-name) and pathogen will handle everything for us – awesome! Most plugin developers set-up their folders to work nicely with pathogen to make life easy.
mkdir -p ~/.vim/autoload ~/.vim/bundle curl -so ~/.vim/autoload/pathogen.vim https://raw.githubusercontent.com/tpope/vim-pathogen/master/autoload/pathogen.vim
Section 4: Installing plugins
The first plugin we will use is called powerline and it adds features and makes a better looking status line.
#install powerline plugin cd ~/.vim/bundle git clone git://github.com/Lokaltog/vim-powerline.git
The next plugin allows code folding to make it easier to look through long blocks of code. Its simple to use, just type f to collapse a section of code of F to collapse it all. Type again to reverse the folding – sweet as.
# install folding flugin us f to fold block of F to fold all mkdir -p ~/.vim/ftplugin wget -O ~/.vim/ftplugin/python_editing.vim http://www.vim.org/scripts/download_script.php?src_id=5492
The next one installs ctrlp which allows nice fuzzy file searches from directly inside vim.
#install ctrlp cd ~/.vim/bundle git clone https://github.com/kien/ctrlp.vim.git
Install the jedi plugin which allows awesome auto-completion of commands and imports using [ctl][space].
#install jedi plugin git clone --recursive https://github.com/davidhalter/jedi-vim.git
Install syntastic is another awesome plugin that does syntax checking and will check your code for compliance with PEP8.
#install syntastic git clone https://github.com/scrooloose/syntastic.git
Forget tabbing into the terminal to stage/commit/merge etc just install git support with fugitive.
# install git support cd ~/.vim/bundle git clone git://github.com/tpope/vim-fugitive.git vim -u NONE -c "helptags vim-fugitive/doc" -c q
So that is it! Your editor should look something like this:
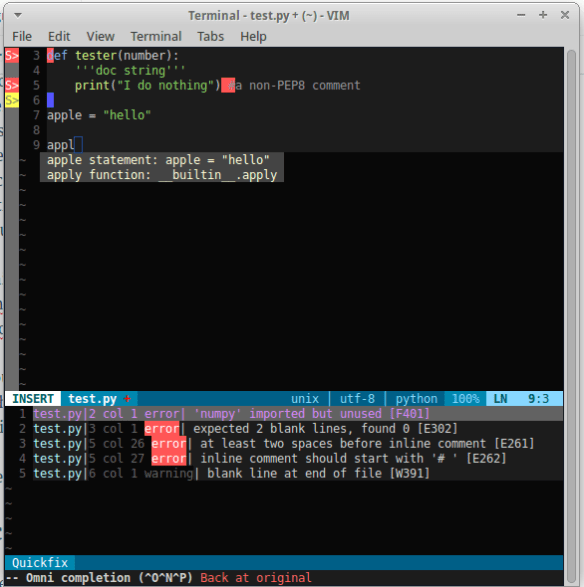
Well that’s the install. Next post will be the shortcuts I use all the time and some that I want to learn as a bookmark for myself. Otherwise checkout the links to the plugins for detailed descriptions of what they do. But now you should be up and running with a pretty nice looking IDE!